

新型コロナウィルスってご存知でしょうか。
僕はあまりテレビを見ないのでよく知らないのですが、今回は流行っているらしい新型コロナウィルスをpythonを使って解析してみようというお話です。(解析といっても、大したことはしませんが...。)
Biopythonの使い方を新型コロナウィルス(SARS-CoV-2 )を題材に学んでいきます。
Biopythonでできること
Biopythonを使えばこんなことができます。
- 様々なバイオインフォマティクスファイルをpythonで利用
以下の形式をサポートしています
Blast(スタンドアローン版とWeb版の両方)、FASTA、Clustalw、GenBank
PubMed、Medline、EnzymeやPrositeなどのExPASyファイル、'dom'および 'lin'ファイルを含むSCOP
UniGene、SwissPro
- バイオインフォマティクスプログラムへのインターフェースとして利用
NCBI(Blast)のスタンドアロン版、Clustalwアラインメントプログラム、EMBOSSコマンドラインツールをBiopython経由で扱うことができます。 - シーケンスに対して、翻訳、転写、重量計算などの処理を行う事ができる
- k近傍法、単純ベイズ分類器、またはサポートベクターマシンを使用したデータの分類
新型コロナウィルスのシーケンスを見てみよう
それでは、本題です。新型コロナウィルス(SARS-CoV-2)のアミノ酸配列を見てみましょう。
Biopythonをインストールすることを忘れずに
pip install biopythonデータベースからシーケンス情報を取得
まず、Biopythonを用いて新型コロナウィルスのシーケンスを取得する必要があります。
今回はこちら(NCBI BLAST)のデータベースからシーケンスの取得を行います。
新型コロナウィルス(SARS-CoV-2)のシーケンス情報は以下から取得します。
https://www.ncbi.nlm.nih.gov/nuccore/NC_045512
以下のように記述するだけでseqdataにシーケンス情報が格納されます。
from Bio import Entrez
from Bio import SeqIO
from collections import Counter
from Bio.SeqUtils import *
#ルール、マナーとして使うときはEメールを入れる
Entrez.email = "xxx@xxxxx.xx.xx"
handle = Entrez.efetch(db='Nucleotide', id='NC_045512', rettype='gb')
seqdata = SeqIO.read(handle, 'genbank')Entrez.emailには、データベースから情報を取得する際にルール・マナーとしてEmailを入力しておきます。(記載しなくても良いのですが、念の為ということで..)
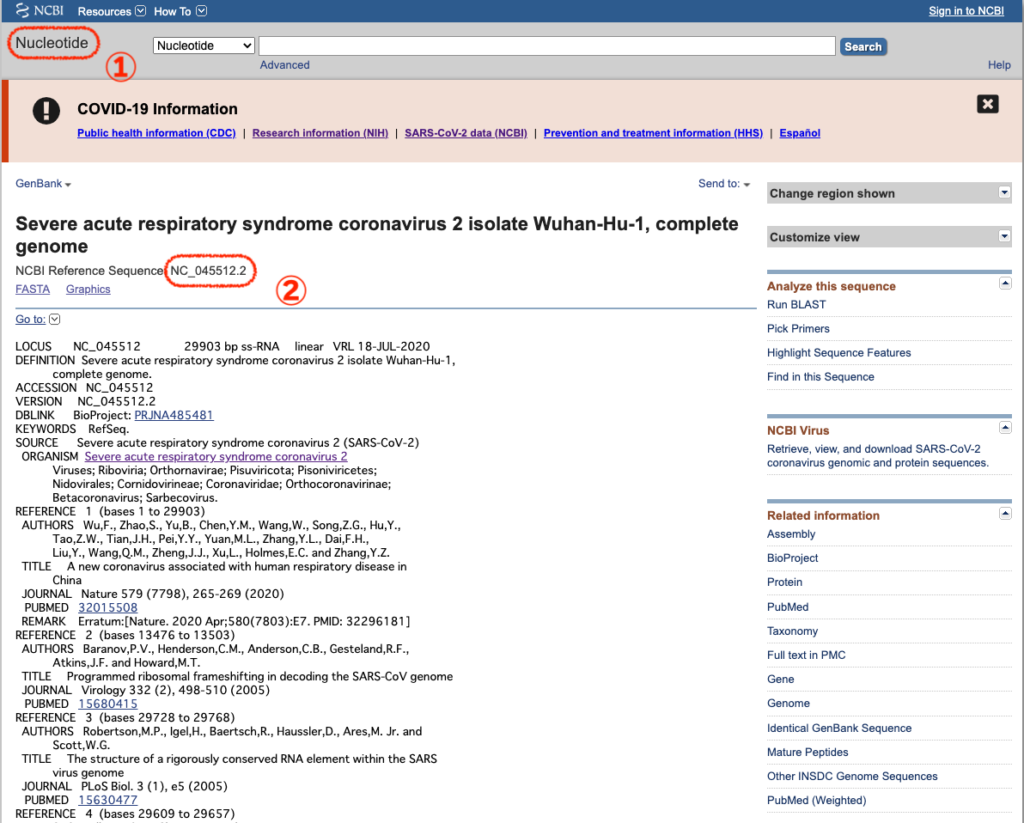
db、idはNCBI Blastのページから確認します。
以下の①がdb情報(データの種類)、②がid情報(データの管理番号)となります。idは小数点以下は切り捨てます。NC_045512.2の場合NC_045512です。rettypeは固定値gbを使います
試しに取得したシーケンスデータを表示してみます。
print(seqdata)
>>>
ID: NC_045512.2
Name: NC_045512
Description: Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
Database cross-references: BioProject:PRJNA485481
Number of features: 57
/molecule_type=ss-RNA
/topology=linear
/data_file_division=VRL
/date=18-JUL-2020
/accessions=['NC_045512']
/sequence_version=2
/keywords=['RefSeq']
/source=Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
/organism=Severe acute respiratory syndrome coronavirus 2
/taxonomy=['Viruses', 'Riboviria', 'Orthornavirae', 'Pisuviricota', 'Pisoniviricetes', 'Nidovirales', 'Cornidovirineae', 'Coronaviridae', 'Orthocoronavirinae', 'Betacoronavirus', 'Sarbecovirus']
/references=[Reference(title='A new coronavirus associated with human respiratory disease in China', ...), Reference(title='Programmed ribosomal frameshifting in decoding the SARS-CoV genome', ...), Reference(title='The structure of a rigorously conserved RNA element within the SARS virus genome', ...), Reference(title="A phylogenetically conserved hairpin-type 3' untranslated region pseudoknot functions in coronavirus RNA replication", ...), Reference(title='Direct Submission', ...), Reference(title='Direct Submission', ...)]
/comment=REVIEWED REFSEQ: This record has been curated by NCBI staff. The
reference sequence is identical to MN908947.
On Jan 17, 2020 this sequence version replaced NC_045512.1.
Annotation was added using homology to SARSr-CoV NC_004718.3. ###
Formerly called 'Wuhan seafood market pneumonia virus.' If you have
questions or suggestions, please email us at info@ncbi.nlm.nih.gov
and include the accession number NC_045512.### Protein structures
can be found at
https://www.ncbi.nlm.nih.gov/structure/?term=sars-cov-2.### Find
all other Severe acute respiratory syndrome coronavirus 2
(SARS-CoV-2) sequences at
https://www.ncbi.nlm.nih.gov/genbank/sars-cov-2-seqs/
COMPLETENESS: full length.
/structured_comment=OrderedDict([('Assembly-Data', OrderedDict([('Assembly Method', 'Megahit v. V1.1.3'), ('Sequencing Technology', 'Illumina')]))])
Seq('ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT...AAA')いろんな情報が格納されていますね。ここから欲しいデータを取得しながら解析を行っていきます。
塩基配列の取得
では先程取得してきたシーケンス情報から塩基配列を取得してきます。
塩基配列の取得は以下のようにして取得します。
base_seq = seqdata.seqこれでbase_seqには「ATTAAAGGTTTATACCTTCCCAGGTAACAAA...」のような情報が格納されます。
シーケンス情報のプロパティは以下のようになっています。
seq 配列データid 管理番号情報name LOCUS などに相当する配列の名前description 配列の補足説明letter_annotations 付加情報annotations 付加情報 マップ型features SeqFeature オブジェクト(エクソン、イントロンなどのアノテーション情報など)dbxrefs 文献情報
管理番号情報を取得したい場合はseqdata.idとかで取得すればいいわけですね。
アミノ酸配列への変換
塩基配列からアミノ酸配列へ変換していきます。
イントロンの部分を取得し、アミノ酸配列へ変換してみましょう。
イントロンの塩基配列番号の取得はシーケンス情報のfeaturesから取得してきます。
ft = seqdata.features[3]そして、塩基配列のイントロン部分をアミノ酸配列へ変換します。
intron_seq = seqdata.seq[ft.location.start:ft.location.end]
amino_seq = intron_seq.translate()イントロンの開始はft.location.start、終了はft.location.endで取得できます。
そこからtranslate()で翻訳を行い、amino_seqにアミノ酸配列が格納されます。
試しに出力してみましょう。
print(amino_seq)
>>>
MESLVPGFNEKTHVQLSLPVLQVRDVLVRGFGDSVEEVLSEARQHLKDGTCGLVEVEKGVLPQLEQPY...いかがでしょうか。これで新型コロナウィルスのアミノ酸配列が簡単に取得できました。
ですが、実はこの配列はNCBI Blastに管理されている情報なのでBiopythonを使わずとも、コピペで簡単に取得できるんですけどね...。データベースって偉大!!
研究の過程で独自のバイオシーケンスなどを扱う際はこのようにして簡単にアミノ酸に変換したり、計算を行う事もできますので便利です。
GC含有量を調べてみる
おまけです。新型コロナウィルスのGC含有量も簡単に調べることもできます。
gc_rate = GC(amino_seq)
print(gc_rate)
>>> 37.728854592222724GC含有量は約37.73%でした。
基本的にGC含有量が高い(GCリッチ)とPCRを行いにくいという特性がありますが、新型コロナウィルスはGC含有量が高くないためPCRを行いやすいのです。だからPCR検査で調べることができるんですね。
...というようにPythonを使ってバイオ系のデータを扱ったり分析を行ったりすることができるんです。
もっと、Biopythonについて調べてみようと思います。次回の記事もお楽しみに

コメント